I disordini immuno-mediati associati alla sottoclasse di anticorpi IgG4, e riuniti sotto la dicitura generica di IgG4 related diseases (IgG4-RD), racchiudono patologie caratterizzate da disordini antigene-specifici, che interessano esclusivamente il sistema nervoso centrale o periferico, definiti come IgG4-neurologic disorders (IgG4-ND). Tale neuro-autoimmunità sarebbe sostenuta dalla produzione di autoanticorpi IgG4 (IgG4-Ab) che esercitano un effetto patogenetico diretto attraverso differenti meccanismi, quali:
- IgG4 Ab diretti contro antigeni neurali anti-MuSK nella Miastenia Gravis.
- IgG4 Ab contro proteine nodali/paranodali che alterando l’integrità del nodo di Ranvier possono determinare un quadro di poliradicolopatia demielinizzante infiammatoria cronica (CIDP).
- IgG4 Ab contro l’antigene LGI1 (leucine-rich, glioma-inactivated-1) o contro il complesso Caspr2 (juxtaparanodal contactin-associated protein-like 2), come in alcune forme di encefalite limbica associata a crisi epilettiche (LGI1), sindrome di Morvan e neuromiotonia (Caspr2).
- IgG4 Ab contro IgLON5, una molecola di adesione espressa nel sistema nervoso, determinanti una rara sindrome caratterizzata da instabilità̀ posturale, discinesie cranio-faciali, disturbi del sonno e deterioramento cognitivo.
La peculiarità dei disordini correlati ad IgG4 è la scarsa risposta alle immunoglobuline intravenose (IVIg). Tali disordini, a differenza delle più frequenti forme mediate da autoanticorpi IgG1, presentano un’inadeguata e variabile risposta agli steroidi e alla plasmaferesi, ma una buona risposta alle terapie umorali anti CD-20, come il Rituximab. Le IgG4 sono la sottoclasse di IgG meno comune, comprendono infatti solo il 5% delle IgG totali, e sono monovalenti. Aumentano, in genere, dopo esposizione cronica ad alte dosi di allergeni e si suppone che la loro produzione sia connessa al fenomeno immunologico della tolleranza periferica. A differenza delle altre classi di IgG, non possono attivare la cascata del complemento e non si legano ai recettori inibitori FcγRIIB. Per tali ragioni, non attivano in maniera adeguata la risposta cellulo-mediata né quella mediata dal complemento, sfuggendo all’azione diretta delle IVIg e delle altre immunoterapie convenzionali.
È importante per il neurologo conoscere questi disordini specifici IgG4-correlati, poiché nonostante si possano talora presentare con quadri clinici peculiari, generalmente si manifestano con quadri simili a quelli dei disturbi neurologici autoimmuni IgG1 mediati, come nelle forme classiche di CIDP, e ciò può portare ad un ritardo nel loro riconoscimento e nell’inizio della terapia adeguata. Inoltre, non è raro che nel corso della malattia, specie nella CIDP, si manifestino switch isotipici da IgG1 o IgG3 verso IgG4, con conseguente sviluppo di “resistenza” alle IVIg. Queste osservazioni assumono una particolare rilevanza per la valutazione e la gestione delle poliradiculoneuropatie infiammatorie croniche, dal momento che è stato recentemente dimostrato che la patogenesi, l’istopatologia, la presentazione clinica e la risposta terapeutica differiscono a seconda che il processo autoimmune sia IgG4 mediato o meno.
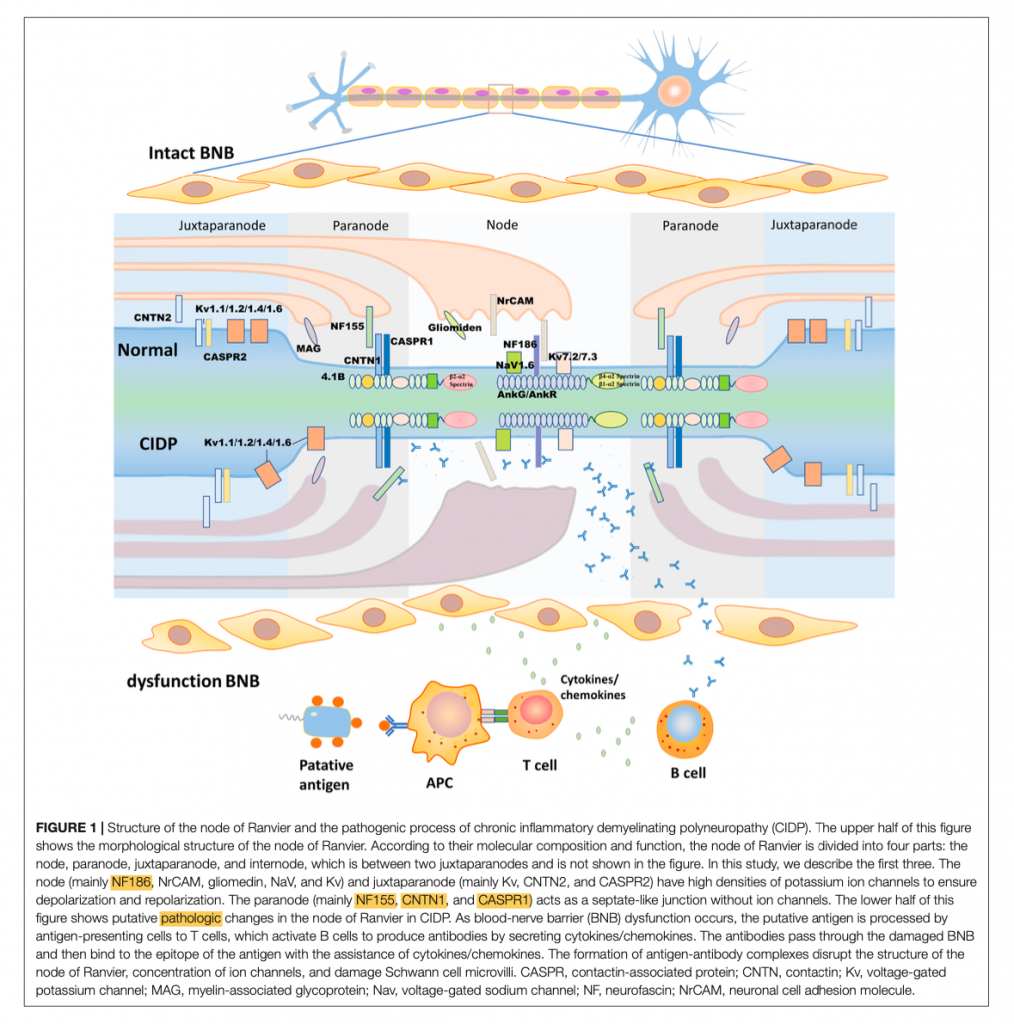
Per comprendere questa differenza è opportuno ricordare la composizione delle fibre mieliniche e la base strutturale della componente saltatoria dell’impulso nervoso a livello del sistema nervoso periferico (SNP). Nell’uomo, buona parte delle fibre del SNP sono rivestite da mielina, grazie alla funzione delle cellule di Schwann. Tale guaina mielinica aumenta la resistenza transmembrana e ne riduce la capacità elettrica, e può essere divisa in 4 parti: nodo di Ranvier propriamente detto, paranodo, juxtaparanodo e internodo (fig.1). Il nodo di Ranvier si colloca nel gap che si viene a formare tra due segmenti di guaina mielinica, ed è ricoperto da strati di cellule di Schwann e microvilli. A questo livello, sul versante assonale sono presenti canali ionici per il sodio e per il potassio, proteine del citoscheletro e una molecola di adesione cellulare, la neurofascina 186 (NF186); sul versante della cellula di Schwann sono invece espresse le molecole di adesione cellulare neuronale (NrCAM), che secernono fattori favorenti il processo di concentrazione di NF186 e di assemblaggio del nodo. Il paranodo è una barriera strutturale che comprende principalmente tre molecole: CNTN1 e Caspr1 sul versante assonale, e la neurofascina 155 (NF155), espressa sulla cellula di Schwann.
Gao et al. Front. Mol. Neurosci., 2021
Negli ultimi anni, in un limitato gruppo di pazienti con CIDP che avevano in comune i meccanismi immunopatologici, la manifestazione clinica, la risposta terapeutica e che differivano da quelli della tipica CIDP, sono stati descritti autoanticorpi rivolti contro le molecole di adesione del complesso paranodale, in particolare contro NF155, CNTN1 e Caspr1, e contro NF186, molecola assonale a livello del nodo di Ranvier. Le IgG4 sono l’isotipo predominante di questi autoanticorpi. Ciò ha portato alla definizione di nodopatia o paranodopatia autoimmune e alla sua introduzione nei criteri diagnostici della CIDP secondo la European Academy of Neurology/Peripheral Nerve Society.
Tali autoanticorpi riescono ad espletare la loro attività patogenica sfruttando le aree in cui la Blood-Nerve-Barrier (BNB) è deficitaria e più permeabile. In caso di infezione, traumi o alterazioni del sistema immunitario, la produzione e il richiamo di citochine e cellule dell’immunità determina la distruzione delle tight junctions e modifica la permeabilità della BNB, rappresentando il primo step nella cascata patologica della CIDP. Infatti, le molecole di interazione tra assone e cellula di Schwann, come NF, CNTN1 e Caspr1, risultano così accessibili al sistema immunitario e vengono espresse dalle cellule presentanti gli antigeni ai linfociti T, con conseguente attivazioni di linfociti B e produzione di autoanticorpi. Questi, attraversano la BNB e si legano alle sopracitate molecole di adesione target, con la formazione di complessi antigene-anticorpo che bloccano l’interazione tra assone e cellula di Schwann e determinano la disgregazione delle strutture nodali e paranodali. A differenza delle più comuni forme di CIDP (sieronegative o non antigene specifiche), che sono caratterizzate da una patogenesi cellulo-mediata, con una prevalenza di macrofagi, e in misura minore di linfociti T, che esitano in un quadro demielinizzante, le forme sieropositive a IgG4 determinano il deficit di conduzione saltatoria attraverso il blocco funzionale dell’interazione tra le componenti gliali e assonali a livello del nodo e del paranodo. Ciò è evidente anche allo studio istopatologico: le tipiche alterazioni della CIDP sono rappresentate da aree a “bulbo di cipolla” di demielinizzazione e remielinizzazione, e da un infiltrato perivascolare di cellule infiammatorie. Nelle forme sieropositive antigene-specifiche invece, si riscontra il distacco tra l’assolemma e il loop mielinico terminale a livello gliale, associato ad un anomalo slargamento dello spazio periassonale, come per distruzione delle giunzioni paranodali, in assenza di aree di demielinizzazione e di infiltrato cellulare perivascolare; inoltre, talora è presente severa perdita e degenerazione assonale.
Tali distinte entità patogenetiche differiscono anche dal punto di vista clinico. La presentazione clinica tipica della CIPD è caratterizzata da una neuropatia periferica simmetrica, a predominanza motoria, con ipostenia prossimale e distale, disturbi della sensibilità specialmente a carico della pallestesia e della batiestesia, areflessia, interessamento dei nervi cranici, sintomi autonomici, e meno comunemente dolore neuropatico. La presenza di dissociazione albumino-citologica nel liquor e l’edema simmetrico delle radici nervose alla RM supportano la diagnosi. Manifestazioni atipiche includono disturbi di forza o di sensibilità isolati, o la sindrome di Lewis-Sumner (neuropatia demielinizzante multifocale con blocchi di conduzione). La prevalenza aumenta con l’età e vi è una predominanza maschile. In genere viene riferita in anamnesi una infezione gastro-intestinale o del tratto respiratorio superiore, circa 4-6 settimane prima dell’esordio dei sintomi, mentre altre volte viene preceduta da quadri di epatite virale o da una vaccinazione.
Le nodopatie e paranodopatie autoimmuni presentano delle specifiche caratteristiche anche in base al tipo di autoanticorpo coinvolto nel processo patogenetico, ma è importante sottolineare che tali differenze si apprezzano qualora l’isotipo immunoglobulinico predominante sia IgG4: i pazienti con autoanticorpi di tipo IgG1 o IgG3 contro le strutture nodali o paranodali, infatti, non mostrano caratteristiche cliniche distinte rispetto alle forme di CIDP tipica.
Il fenotipo clinico delle forme anti-NF155 è caratterizzato da un esordio più precoce (intorno ai 20- 30 anni), subacuto e più severo, con interessamento motorio a predominanza distale, associato a tremore a bassa frequenza e ad ampie scosse e ad atassia cerebellare; talora vi può essere il coinvolgimento dei nervi cranici, in particolare con paralisi faciale. Peraltro, recentemente è stata rilevata una forte associazione con l’HLA DRB1*15, fattore di rischio costitutivo per le forme di CIDP anti-NF155.
Gli anticorpi anti-CNTN1 determinano un quadro ad esordio subacuto o acuto rapidamente progressivo di severa neuropatia motoria e sensitiva, con interessamento assonale precoce, atassia sensitiva e tremore, seppur con frequenza minore rispetto ai pazienti anti-NF155 +. Tipica delle forme anti-CNTN1 è la possibilità di una concorrente presenza di una nefropatia a IgG4, ed esordiscono ad un’età più avanzata rispetto alle forme anti-NF155.
Più rare e meno conosciute sono le forme anti-Caspr1 e anti-NF186: le prime si distinguono per la variabile presenza di importante dolore neuropatico, mentre le seconde per una spiccata atassia sensitiva, blocchi di conduzione e interessamento dei nervi cranici.
I pazienti con anticorpi contro i componenti del nodo e del paranodo mostrano una scarsa risposta a IVIg, con l’eccezione delle forme anti-NF186, probabilmente perché le IgG4 non rappresentano l’isotipo dominante e perché la NF186 ha una localizzazione più accessibile alle Ig in circolo. La risposta ai corticosteroidi è variabile e anche in questo caso i pazienti anti-NF186 sembrano essere più sensibili. In generale, è stata osservata una buona risposta a Rituximab, in particolare per le forme anti-NF155 e anti-CNTN1. L’espansione delle cellule B attivate, l’evidenza di un ruolo patogenetico diretto, e l’efficacia clinica delle terapie depletive, supportano il Rituximab come trattamento di prima scelta in questo tipo di pazienti. Alcuni studi hanno inoltre dimostrato che i titoli sierici di autoanticorpi possono rappresentare un buon biomarker per valutare l’attività di malattia e la risposta terapeutica: nei pazienti trattati con Rituximab il titolo di IgG4 si riduce di più del 90%, e ciò suggerisce una correlazione fra il titolo anticorpale e le riesacerbazioni. Secondo alcuni autori è preferibile ripetere le infusioni qualora occorrano recidive cliniche, altri propongono una terapia con Rituximab 2 g ogni 6 mesi o 1 g ogni 3 mesi, al fine di garantire la stabilità della malattia.
Negli ultimi anni le caratteristiche delle nodo-paranodopatie autoimmuni vanno sempre più delineandosi, al punto che, secondo alcuni autori, dovrebbero essere considerate come un’entità patologica indipendente e non come un sottotipo di CIDP e potrebbero essere classificate come nodo-paranodopatie croniche CIDP-like. La ricerca di anticorpi contro NF155, CNTN1, Caspr 1 e NF186, con conseguente determinazione dell’isotipo di IgG nei casi sieropositivi, dovrebbe essere parte integrante dell’iter diagnostico delle neuropatie infiammatorie croniche, in modo da guidare e non ritardare l’approccio terapeutico più adeguato.
Dott. Giuseppe Piga
Università degli Studi di Cagliari
giuseppe.piga92@gmail.com
Bibliografia:
- Marinos C. Dalakas, IgG4-Mediated Neurologic Autoimmunities. Neurol Neuroimmunol Neuroinflamm 2022;9e1116.
- Gao et al. Impact of Neurofascin on Chronic Inflammatory Demyelinating Polyneuropathy via Changing the Node of Ranvier Function: A Review. Front. Mol. Neurosci. 2021,14:779385.
- Van den Bergh PYK, Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second Revision. J Peripher Nerv Syst. 2021;26:242–268.
- Tang L, Tang X et al., Distinguish CIDP with autoantibody from that without autoantibody: pathogenesis, histopatology, and clinical features. J Neurol 2021 Aug;268(8):2757-2768.
- Cortese A, Lombardi R, Devaux J, Franciotta D et al., Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP. Neurol Neuroimmunol Neuroinflamm 2020;7:e639.
- Martìn-Aguilar L, Querol L, et al. Clinical and laboratory features in anti-NF155 autoimmune nodopathy. Neurol Neuroimmunol Neuroinflamm 2022;7:e1098.