Da Silber MH. Autoimmune sleep disorders. Handb Clin Neurol. 2016;133:317-26. doi: 10.1016/B978-0-444-63432-0.00018-9. PMID: 27112685.
In questo articolo affronteremo le encefaliti autoimmuni e/o paraneoplastiche associate alla presenza di autoanticorpi, in cui i disturbi del sonno rappresentano spesso la manifestazione clinica preponderante. In queste condizioni patologiche gli autoanticorpi identificati sono stati in alcuni casi ritenuti implicati nella patogenesi del quadro clinico, in altri rappresenterebbero più verosimilmente un epifenomeno del processo disimmune in atto. Va sottolineato che alcuni disturbi del sonno “idiopatici” sono considerati, oggi, a patogenesi autoimmune, su tutte la Narcolessia. Queste patologie, però, non saranno argomento di questa trattazione.
Anticorpi contro il complesso del canale del potassio voltaggio dipendente
Gli anticorpi diretti contro i complessi del canale del potassio voltaggio-dipendente (VGKC) sono un gruppo di autoanticorpi il cui target è rappresentato da molecole di superficie che interagiscono con la subunità Kv1 dei VGKC in corrispondenza o nelle prossimità delle sinapsi. Nell’ambito di tale complesso sono state identificate almeno due proteine: la “contactin-associated protein-2” (CASPR2), situata a livello dei paranodi e ricca in leucina, e la “glioma-inactivated protein 1” (LGI1), situata a livello della sinapsi e particolarmente rappresentata nel neuropilo dell’ippocampo. Questi auto-anticorpi sono stati associati a due principali disordini neurologici: la sindrome di Morvan e l’encefalite limbica.
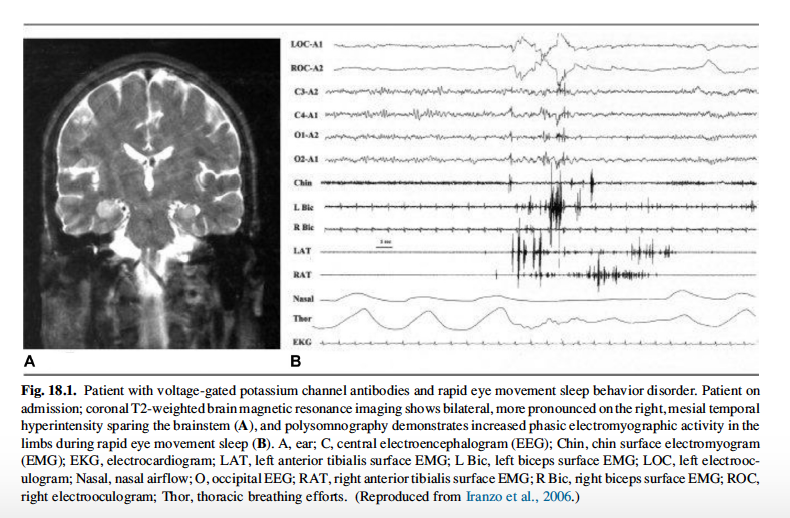
La sindrome di Morvan è una rara malattia che coinvolge sia il sistema nervoso periferico che quello centrale. L’ipereccitabilità del sistema nervoso periferico si manifesta con miochimie, fascicolazioni e neuromiotonia. Un’insonnia profonda, un’encefalopatia dall’andamento fluttuante con allucinazioni e un disturbo disautonomico caratterizzano l’interessamento del sistema nervoso centrale. Il 90% circa dei casi riportati sono di sesso maschile. In circa la metà dei casi la malattia è sottesa dalla presenza di una neoplasia, in particolare timomi. Entrambi gli autoanticorpi, CASPR2 e LGI1, sono stati rilevati in pazienti con sindrome di Morvan, nonostante i CASPR2 si mostrino a più alti titoli. L’insonnia è la manifestazione clinica preminente e più severa del quadro sindromico. La polisonnografia e l’actigrafia del polso spesso mostrano una marcata riduzione del tempo totale di sonno o la sua completa assenza. Il sonno REM, il sonno a onde lente e la fase N2 sono marcatamente ridotti o assenti. Non sono inoltre rilevabili i fusi del sonno o i complessi K. L’attività elettroencefalografica di fondo mostra prevalentemente un’attività theta disorganizzata. Quando il sonno REM è presente, l’atonia muscolare tipica è assente, per cui il comportamento notturno si caratterizza per agitazione e sogni “partecipati”, configurando uno stato noto come agrypnia excitata, similmente a quanto accade nell’insonnia familiare fatale. Nonostante il sonno notturno sia severamente e intensamente compromesso, non è ravvisabile una severa ipersonnia diurna. Le indagini di neuroimaging nella maggior parte dei casi non sono informative. Il substrato anatomofisiologico dei disturbi del sonno è sconosciuto ma è stato postulato un coinvolgimento della circuiteria talamo-limbica. L’insonnia è solitamente refrattaria al trattamento con ipnotici convenzionali. Alcuni scarsi report segnalano un beneficio dal trattamento con oppiacei. Al contrario, un marcato miglioramento dei disturbi del sonno si è riscontrato con l’immunoterapia (plasmaferesi, immunoglobuline, steroidi e ciclofosfamide).
L’encefalite limbica associata ad autoanticorpi contro il complesso VGKC è caratterizzata sul piano clinico da crisi epilettiche, confusione e compromissione della memoria e sul piano neuroradiologico da un aumento del segnale in T2 nella porzione mediale dei lobi temporali. In questa forma risultano più frequentemente implicati gli anticorpi anti-LGI1. Non è frequente che la condizione sia sottesa da neoplasie, anche se sono stati riportati alcuni casi associati ad adeoncarcinoma della prostata e del colon. I disturbi del sonno non sono dissimili da quelli descritti per la sindrome di Morvan: disturbi del comportamento del sonno REM, sogni “partecipati” e profonda insonnia in associazione ad agrypnia excitata. In qualche caso può essere viceversa presente ipersonnia. Le manifestazioni neurologiche e del sonno in alcuni i pazienti si sovrappongono tra sindrome di Morvan ed encefalite limbica, velando la distinzione tra le due condizioni. I disturbi del sonno nell’encefalite limbica sono spesso molto reattivi all’immunoterapia.
Da Silber MH. Autoimmune sleep disorders. Handb Clin Neurol. 2016;133:317-26. doi: 10.1016/B978-0-444-63432-0.00018-9. PMID: 27112685.
Anticorpi anti-acquaporina 4
La neuromielite ottica (NMO) è una malattia demielinizzante caratterizzata da neurite ottica recidivante e mielite longitudinalmente estesa. Sono stati descritti un piccolo numero di casi di narcolessia associata alla presenza di NMO-IgG e lesioni iperintense ipotalamiche bilaterali. Di questi casi tutti, tranne uno (Carlander et al., 2008), erano giapponesi, una popolazione nota per avere una prevalenza relativamente più alta di NMO a confronto con popolazioni di origine europea. Ulteriori pazienti con coinvolgimento ipotalamico ed eccessiva sonnolenza diurna di altre etnie sono stati descritti, ma senza un test adeguato a caratterizzarli come affetti da narcolessia (Poppe et al., 2005; Viegas et al., 2009; Samart e Phanthumchinda, 2010).
I pazienti con narcolessia ben definita hanno mostrato a quadro clinico relativamente coerente. In 5 pazienti su 8, l’ipersonnia ha preceduto altre manifestazioni di NMO, mentre i restanti 3 hanno presentano un quadro diencefalico isolato. In un paziente, l’ipersonnia ha presentato carattere ricorrente. La cataplessia, solitamente associata a Narcolessia di tipo 1, non era presente in nessun paziente. Il disturbo del sonno si associava perlopiù ad altri segni di disfunzione ipotalamica, tra cui: ipotermia, disautonomia e sindrome da inappropriata secrezione di ormone antidiuretico.
La valutazione dei livelli di ipocretina-1 nel liquor ha dimostrato concentrazione bassa (<110 pg / mL) in 3 pz e intermedia (110-200 pg / mL) in 5 su 8 casi. Il test di latenza del sonno multiplo ha mostrato periodi di sonno ad esordio REM (sleep-onset REM sleep periods; SOREMPS) in tutti e 3 i casi in cui il test è stato eseguito. Nei 2 pazienti studiati per i polimorfismi HLA, il test non ha mostrato gli antigeni tipici associati alla narcolessia idiopatica. La somministrazione di steroidi per via endovenosa e per via orale ha visto un miglioramento della sonnolenza in 4 su 5 pazienti, associandosi ad aumento dei livelli di ipocretina-1 nel liquido cerebrospinale e risoluzione o miglioramento dell’iperintensità ipotalamica in RM. La risposta alla terapia così come i livelli intermedi di ipocretina-1 nel liquido cerebrospinale nella maggior parte dei casi, suggeriscono che la disfunzione ipotalamica è spesso parziale e potenzialmente reversibile, probabilmente associata a disfunzione neuronale piuttosto che a morte cellulare.
Anticorpi contro il recettore NMDA
Gli anticorpi anti-NMDAR sono stati associati clinicamente a distinte forme di encefalite autoimmune caratterizzate precocemente da sintomi neuropsichiatrici, quali agitazione, pensiero delirante e allucinazioni, sviluppo di epilessia, disturbo della memoria, seguiti da disautonomia, discinesie orofacciali e degli arti e diminuzione dei livelli di coscienza. Si tratta di una condizione che più frequentemente si verifica nell’infanzia o nella giovane età adulta, coinvolgendo in circa il 90% dei casi il sesso femminile. E’ possibile osservare una genesi paraneoplastica nel 20-60% dei pazienti, il tumore più comunemente implicato è il teratoma ovarico. Il trattamento della neoplasia sottostante e l’immunoterapia hanno portato a un miglioramento in molti pazienti. I disturbi del sonno nel contesto delle encefaliti anti-NMDA non sono ancora ben caratterizzati. Sulla base delle attuali evidenze sembra che, in una fase non iniziale di malattia, sia riscontrabile un’ipoventilazione centrale nel 66% dei pazienti, molti dei quali richiedono un ventilatore di supporto per periodi di 2-40 settimane. L’apnea notturna centrale è stata segnalata in 2 pazienti. Grave insonnia, non responsiva alla terapia, è stata segnalata durante la fase acuta della malattia in 2 pazienti (Poloni et al., 2010) e preminenti disturbi del sonno (ipersonnia e inversione di modelli di sonno) sono stati osservati nel 27% dei pazienti dopo il recupero.
In uno studio su 20 soggetti di età pediatrica che presentavano un fenotipo clinico compatibile con encefalite letargica, tra i 10 bambini positivi per anticorpi NMDAR, 6 riportavano insonnia e 2 inversione del sonno durante il decorso della loro malattia. Nei restanti 10 (negativi per la presenza di anticorpi NMDAR), al contrario, prevaleva una condizione di ipersonnia. Infatti, 5 mostravano ipersonnia, 3 inversione del sonno e solo 1 insonnia (Dale et al., 2009). Ulteriori studi clinici e polisonnografici dei disturbi del sonno in diverse fasi della malattia sono necessari per meglio definire la condizione.
Anticorpi IgLON5
Di recente identificazione è un disturbo del sonno caratterizzato da parasonnie REM e non REM, apnee ostruttive del sonno e stridor associato alla presenza di anticorpi contro gli epitopi extracellulari della proteina IgLON5, una molecola di adesione neuronale dalle funzioni ancora poco conosciute. I sintomi sono progressivi e possono mettere in pericolo la vita del paziente per insufficienza respiratoria da ipoventilazione centrale. Il quadro neurologico consiste di sintomi bulbari (disartria e disfagia), disautonomia (ipersalivazione, iperidrosi, bradiaritmie e urgenza urinaria), atassia della marcia, anomalie del movimento oculare e corea. I disturbi del sonno sono prominenti. In un recente studio su 22 pazienti i disturbi del sonno riscontrati sono stati: apnee notturne nel 95% dei casi, parasonnie nell’86%, stridor nell’83%, insonnia nel 73%, e ipersonnia diurna nel 60%.
In un precedente studio su 8 pazienti che aveva l’obiettivo di caratterizzare il disturbo del sonno di questa patologia, la polisonnografia ha rivelato vocalizzazioni semplici e complesse, movimenti nella fase N2, intrusioni della fase REM nella fase N2 del sonno e tono muscolare aumentato nel sonno REM, mentre lo stadio N3 risultava normale. Erano presenti movimenti periodici degli arti in veglia e in sonno e l’efficienza e il tempo totale di sonno risultavano ridotti. Quattro degli 8 pazienti inclusi, hanno sviluppato ipoventilazione neurogena centrale, e 6 sono morti improvvisamente, 2 durante il sonno. L’immunoterapia non ha mostrato benefici in 7 pazienti; uno solo è transitoriamente migliorato, per morire poco dopo. La RM e l’analisi del liquido cefalorachidiano sono risultate normali in tutti i pazienti. I livelli di ipocretina-1 nel liquido cerebrospinale erano normali nei 3 pazienti testati. Tutti i 4 pazienti testati presentavano alleli HLA DQB1 * 0501 e HLS DRB1 * 1001 (rari nella popolazione spagnola, a cui appartenevano). I risultati dell’autopsia in 2 pazienti hanno mostrato la presenza di proteina tau iperfosforilata nei neuroni dell’ipotalamo e a livello del tegmento del tronco cerebrale, compresa la materia grigia periacqueduttale, nuclei peduncolopontini, nucleo magnocellulare e nucleo ambiguo. Non sono stati riportati inclusioni di a-sinucleina o elementi di neuroinfiammazione. Queste osservazioni hanno sollevato non pochi interrogativi sul ruolo degli anticorpi IgLON5 in questa condizione: se siano causativi o piuttosto un’epifenomeno del sottostante processo neurodegenerativo. La presenza di patologia tau, l’assenza di alterazioni infiammatorie nel liquor, alla risonanza magnetica o all’autopsia e la mancanza di risposta all’immunoterapia screditerebbero l’ipotesi autoimmune. Tuttavia, il modello HLA visto nei 4 pazienti suggerisce la possibilità di una predisposizione genetica all’autoimmunità. Un’ipotesi più recente teorizza che l’accumulo della proteina tau sia il risultato dell’interferenza, indotta dagli autoanticorpi, nell’interazione di IgLON5 con il citoscheletro intracellulare. In uno scenario di questo tipo la diagnosi e il trattamento precoci diventerebbero cruciali.
Anticorpi MA
Ma1 e Ma2 sono proteine intracellulari ampiamente espresse nel cervello. Gli anticorpi anti-Ma2 sono i più frequenti in pazienti con tumori testicolari, mentre entrambi si trovano in una varietà di tumori, in particolare nel carcinoma del polmone. Sono state descritte varie combinazioni di sindromi paraneoplastiche limbiche, troncoencefaliche e diencefaliche, quest’ultime rappresentanti circa un terzo del totale e spesso associate a disturbi del sonno, su tutti la narcolessia. Dei 22 casi segnalati di narcolessia associata agli anticorpi Ma, tutti presentavano ipersonnia e 6 (27%) cataplessia. Disturbi del comportamento del sonno REM sono stati segnalati in 2 pazienti. La sindrome diencefalica in alcuni casi comprendeva disordini endocrini ipotalamici, inoltre la maggior parte presentava sintomi correlati a coinvolgimento del sistema limbico e del tronco encefalico. I tumori associati includevano neoplasie testicolari, del polmone, e neoplasia tonsillare. La polisonnografia notturna in 4 pazienti ha mostrato ridotta efficienza del sonno, assenza dei fusi del sonno (1 paziente) e sonno a onde lente assente (1 paziente). Il sonno REM senza atonia è stato riportato in 3 pazienti. I test di latenza del sonno hanno mostrato risultati tipici della narcolessia con latenze di sonno medie ridotte e multiple SOREMPS. I livelli di ipocretina-1 nel liquido cerebrospinale sono stati misurati in 12 casi risultando bassi o non rilevabili in tutti, mentre nei 2 pazienti in cui è stato analizzato l’HLA non è stato rilevato la variante caratteristica della narcolessia idiopatica. La concentrazione di proteine nel liquido cerebrospinale, la conta dei globuli bianchi, o entrambi sono risultati aumentati in 4 dei 5 pazienti valutati. La risonanza magnetica cerebrale era anormale in 8 pazienti, ma ha mostrato coinvolgimento diencefalico solo in 4. L’esame istologico dell’ipotalamo in 1 caso ha mostrato infiammazione della materia grigia con cappucci perivascolari, gliosi astrocitaria e infiltrazione di cellule T-CD8+ nel tessuto neurale. Gli effetti dell’immunomodulazione o il trattamento dei tumori primari sono risultati variabili e il piccolo numero di casi segnalati non consente di giungere a conclusioni definitive.
Autoanticorpo nucleare antineuronale di tipo 2 (ANNA-2) (anticorpo anti-Ri)
L’anticorpo paraneoplastico IgG ANNA-2 si lega agli antigeni Nova-1 e Nova-2, proteine nucleari neuronali coinvolte nei processi di splicing alternativo e ampiamente distribuite nel cervello (Yang et al., 1988. La sindrome opsoclono-mioclono è il quadro clinico più tipicamente associato a questi anticorpi, che tuttavia si rintracciano anche in pazienti affetti da encefalite con coinvolgimento tronco-encefalico, cerebellare e midollare. Le neoplasie implicate includono il carcinoma mammario e il carcinoma polmonare sia a piccole che grandi cellule. Una caratteristica di alcuni pazienti è la presenza di stridor.
In una serie di 48 pazienti con anticorpi ANNA-2, 5 (9,6%) presentavano laringospasmo episodico definito e 2 possibile, in 5 pazienti lo stridor era associato con la presenza di distonia di apertura mandibolare. Due pazienti hanno perso coscienza durante gli episodi. Un paziente ha richiesto la tracheotomia, uno l’intubazione d’emergenza e altri 2 il ricovero in unità di cura intensiva. Un paziente è morto durante un episodio di laringospasmo. Il carcinoma della mammella si è rivelato in 5 pazienti, in un paziente il carcinoma a piccole cellule del polmone e in un altro il carcinoma cervicale.
Il laringospasmo è stato riportato anche in un singolo paziente con adenocarcinoma metastatico e polimioclono, associato alla presenza di P / Q e N canale del calcio voltaggio-dipendente e potassio anticorpi del canale, ma non anticorpi ANNA-2.
Referenze
- Cornelius JR, Pittock SJ, McKeon A et al. (2011). Sleep manifestations of voltage-gated potassium channel complex autoimmunity. Arch Neurol 68: 733–738.
- Dale RC, Irani SR, Brilot F et al. (2009). N-methyl-d-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol 66: 704–709.
- Dalmau J, Graus F, Villarejo A et al. (2004). Clinical analysis of anti-Ma2-associated encephalitis. Brain 127:1831–1844.
- Gaig C, Graus F, Compta Y, Högl B, Bataller L, Brüggemann N, Giordana C, Heidbreder A, Kotschet K, Lewerenz J, Macher S, Martí MJ, Montojo T, Pérez-Pérez J, Puertas I, Seitz C, Simabukuro M, Téllez N, Wandinger KP, Iranzo A, Ercilla G, Sabater L, Santamaría J, Dalmau J. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017 May 2;88(18):1736-1743. doi: 10.1212/WNL.0000000000003887. Epub 2017 Apr 5. PMID: 28381508; PMCID: PMC5409845.
- Irani SR, Pettingill P, Kleopa KA et al. (2012). Morvan syndrome: clinical and serological observations in 29 cases.
- Ann Neurol 72: 241–255.
- Iranzo A, Graus F, Clover C et al. (2006). Rapid eye movement sleep behavior disorder and potassium channel antibodyassociated
- limbic encephalitis. Ann Neurol 59: 178–182.
- Liguori R, Vincent A, Clover L et al. (2001). Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 124: 2417–2426.
- Lugaresi E, Provini F, Cortelli P (2011). Agrypnia excitata. Sleep Med 12 (Suppl 2): S3–S10.
- Peter-Derex L, Devic P, Rogemond V et al. (2012). Full recovery of agrypnia associated with anti-Lgi1 antibodies encephalitis under immunomodulatory treatment: a case report with sequential polysomnographic assessment. Sleep Med 13: 554–556.
- Pittock SJ, Lucchinetti CF, Lennon VA (2003). Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol 53: 580–587.
- Pittock SJ, Weinshenker BG, Lucchinetti CF et al. (2006). Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 63: 964–968.
- Pittock SJ, Parisi JE, McKeon A et al. (2010). Paraneoplastic jaw dystonia and laryngospasm with antineuronal nuclear autoantibody type 2 (anti-Ri). Arch Neurol 67: 1109–1115.
Gianmarco Abbadessa
Università degli studi della Campania “Luigi Vanvitelli”
Email: gianmarcoabbadessa@gmail.com