Poco nota e abbastanza rara, la malattia di Perry è stata descritta per la prima volta nel 1975 in una famiglia canadese. Successivamente, nel 2009, è stato individuato il gene responsabile della malattia (DCTN1), portando a un aumento delle diagnosi.
Dal punto di vista clinico si presenta con parkinsonismo, e ha vari punti in comune con la malattia di Parkinson. Dal punto di vista fisiopatologico, invece, fa parte delle proteinopatie TDP-43 relate, e condivide dei meccanismi patogenetici con la Sclerosi Laterale Amiotrofica (SLA) e la demenza fronto-temporale (FTD).
Nel 2011 un consenso di esperti in materia si è riunito a Tokyo per definirne i criteri diagnostici.
Clinica
Dal punto di vista clinico, la malattia di Perry è a tutti gli effetti un parkinsonismo con sintomi atipici. I primi sintomi a svilupparsi, attorno ai 40 anni, sono di natura psichiatrica: una severa depressione, accompagnata da apatia e ideazioni suicidarie, possono rappresentare il motivo del primo contatto medico in questi pazienti.
In seguito si assiste a una riduzione dell’appetito che risulta in un calo ponderale importante (una media di circa -1.5kg al mese), a cui fa seguito l’instaurarsi del parkinsonismo, tendenzialmente simmetrico, che si presenta soprattutto con rigidità. Il tremore, sia a riposo che posturale, è quasi sempre presente; la bradicinesia e l’instabilità posturale si presentano in più di un terzo dei pazienti.
Campanelli d’allarme in pazienti con diagnosi di parkinsonismo che devono far pensare che si possa trattare, invece, di malattia di Perry sono:
- età giovanile (esordio prima dei 50 anni);
- presenza di storia familiare positiva per sintomi simili;
- presenza di manifestazioni cliniche atipiche;
- corso di malattia atipico, particolarmente lungo o, al contrario, breve.
L’ultimo sintomo cardinale della malattia di Perry sono le anomalie di ventilazione, di solito a presentazione tardiva, che rappresentano la principale causa di morte. Le anomalie possono variare dalla tachipnea, alla dispnea, all’ipoventilazione centrale, fino alle apnee; di solito le alterazioni del respiro avvengono di notte, risultando perciò anche in disturbi del sonno come insonnia, sonno interrotto e sonnolenza diurna.
In circa il 25% dei pazienti nel corso della malattia si sviluppano anche deficit cognitivi e, più raramente, fenomeni dispercettivi. Altri sintomi possibili sono rappresentati da segni di disinibizione frontale (iperoralità, riflessi primitivi, deficit esecutivi) e anomalie della motilità oculare (paralisi verticale dello sguardo).
Di norma, l’exitus avviene dopo circa 5 anni dall’esordio dei sintomi.
Fisiopatologia
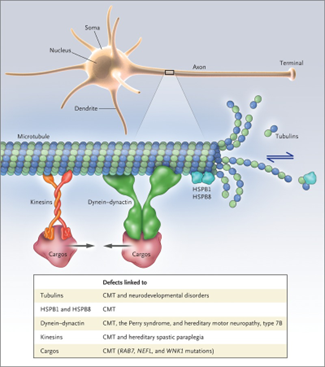
La malattia di Perry è una malattia neurodegenerativa ereditaria a trasmissione autosomica-dominante legata a mutazioni del gene DCTN1 (specialmente dell’esone 2), che si trova sul cromosoma 2 e codifica per la dinactina-1. La dinactina-1 esiste in due versioni (p150-glued e p135) che interagiscono tra loro e con altre proteine, tra cui la dineina, per formare il complesso della dinactina che è indispensabile per il trasporto assonale lungo i microtubuli.
Mutazioni differenti dello stesso gene si ritiene siano implicate anche nello sviluppo di altre patologie neurologiche, tra cui la SLA e la FTD. Un altro punto in comune con queste due patologie appena citate è anche il fatto che, dal punto di vista fisiopatologico, la malattia di Perry si inserisce nel contesto delle proteinopatie TDP-43-relate: sono state infatti riscontrate, in questi pazienti, inclusioni di TDP-43 a livello extrapiramidale e nel tronco encefalico. La corteccia, gli ippocampi e i motoneuroni sono tipicamente risparmiati da queste inclusioni. La spiccata neurodegenerazione a livello del nucleo del rafe dorsale e del bulbo ventrolaterale rappresenta il verosimile substrato delle anomalie respiratorie che questi pazienti sperimentano; i sintomi depressivi, al contrario, si ritengono imputabili alla perdita di neuroni nel locus coeruleus e nell’area tegmentale ventrale. Infine, le alterazioni del sonno dipendono probabilmente dalla perdita di neuroni della formazione reticolare nel tronco encefalico.
Diagnosi
Le analisi ematiche di routine e lo studio del liquor cefalorachidiano non mostrano particolari alterazioni. Anche TC e RM dell’encefalo sono generalmente normali. La PET con FDG e la SPECT mostrano coinvolgimento corticale diffuso. Al DAT-Scan si nota una ridotta captazione di tracciante a livello dello striato bilateralmente e anche alla scintigrafia cardiaca i reperti possono essere compatibili con la presenza di denervazione dopaminergica. È interessante il dato che mostra una ridotta captazione striatale di tracciante anche in pazienti asintomatici carrier di mutazione di DCTN1. La PET con tracciante per la serotonina mostra un ridotto uptake cortico-sottocorticale del tracciante.
Se vi è il sospetto di malattia di Perry diventa fondamentale eseguire una polisonnografia e un’emogasanalisi arteriosa, poiché se questi esami mostrano evidenza di ipoventilazione, il supporto respiratorio può modificare l’aspettativa di vita.
Le analisi genetiche, da proporre ai pazienti ed eventualmente anche ai familiari, mostrano positività per mutazioni del gene DCTN1: sono state identificate 9 mutazioni che si associano a parkinsonismo.
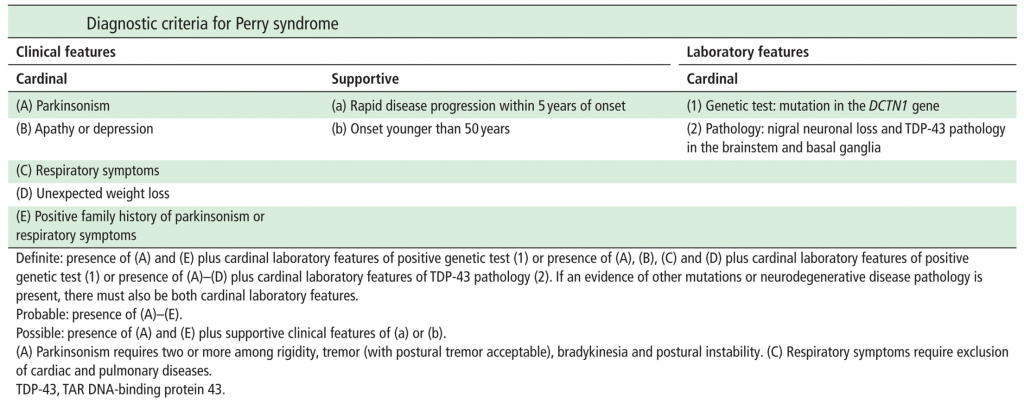
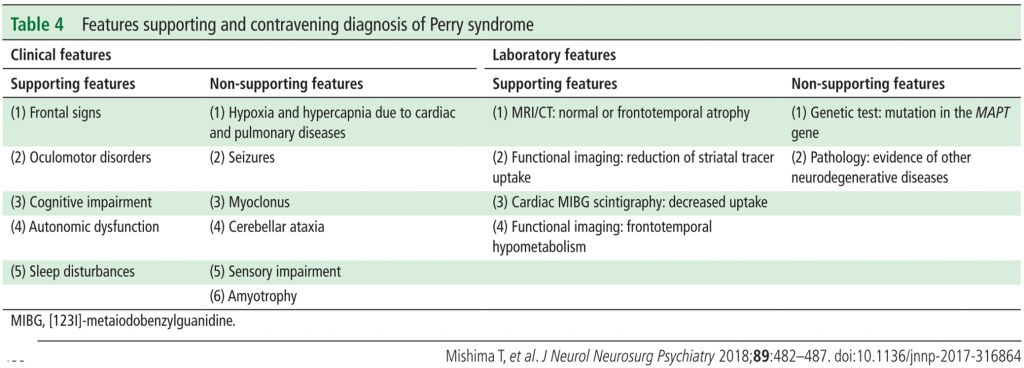
Si rimanda alle tabelle sottostanti per i criteri diagnostici e di supporto alla diagnosi della malattia di Perry (Criteri Diagnostici – il Simposio Internazionale sulla malattia di Perry, Tokyo – 2011).
Trattamento
La maggior parte dei pazienti risponde al trattamento con L-Dopa, almeno inizialmente. Tuttavia, il range di risposta va da “nessuna risposta” a “risposta completa”, quindi è abbastanza variabile.
I sintomi psichiatrici tendono a non essere responsivi al trattamento antidepressivo; è di importanza cruciale far seguire il prima possibile al paziente un percorso di supporto psichiatrico, poiché il tasso di suicidi in questa popolazione è estremamente alto.
Per migliorare l’aspetto nutritivo e far riacquistare peso si possono impostare diete ad alto contenuto calorico e proteico; si è osservato, inoltre, un lieve aumento ponderale anche con il trattamento con amitriptilina. Bisogna ovviamente supplire a tutto ciò di cui il paziente risulta essere carente. Nei casi estremi può essere necessario posizionare un sondino naso-gastrico.
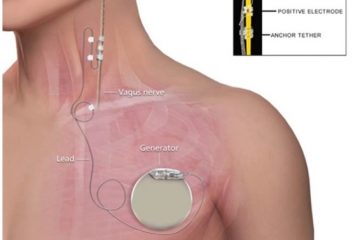
Per la ventilazione, in notturno risulta essenziale un supporto ventilatorio, ad esempio tramite terapia a Pressione Positiva Continua delle vie Aeree (CPAP). Col tempo può diventare necessaria una ventilazione meccanica non invasiva (NIV) continua o addirittura l’intubazione. In alcuni casi è stato impiantato un pace-maker diaframmatico con beneficio.
Adottare tutte queste misure può significativamente migliorare la qualità di vita delle persone affette da malattia di Perry ed è perciò cruciale non ritardarne la diagnosi, imparando a riconoscerla.
Take-home messages
- La malattia di Perry si presenta con 4 sintomi cardine:
- Disturbi psichiatrici (depressione, apatia, ritiro sociale)
- Calo ponderale
- Parkinsonismo (simmetrico)
- Alterazioni respiratorie
- È una malattia genetica a trasmissione autosomica-dominante, con familiari che presentano spesso sintomi simili;
- L’esordio è attorno ai 40 anni, più precoce rispetto agli altri parkinsonismi;
- Il DAT-Scan e la scintigrafia miocardica con MIBG sono positivi per denervazione dopaminergica;
- I pazienti mostrano una buona risposta alla L-DOPA;
- È fondamentale fornire supporto psichiatrico e ventilatorio.
Giorgia Iania
Università degli Studi di Trieste
gi.giorgia.iania@gmail.com
Bibliografia
- Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL. Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol. 1975 Feb;32(2):108-13. doi: 10.1001/archneur.1975.00490440058009. PMID: 1122173.
- Mishima T, Fujioka S, Tomiyama H, Yabe I, Kurisaki R, Fujii N, Neshige R, Ross OA, Farrer MJ, Dickson DW, Wszolek ZK, Hattori N, Tsuboi Y. Establishing diagnostic criteria for Perry syndrome. J Neurol Neurosurg Psychiatry. 2018 May;89(5):482-487. doi: 10.1136/jnnp-2017-316864. Epub 2017 Oct 31. PMID: 29089398; PMCID: PMC5909757.
- Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan M, Kachergus J, Rademakers R, Baker MC, Grantier LL, Sujith OK, Brown L, Calne S, Farrer MJ, Wszolek ZK. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord. 2009 May;15(4):281-6. doi: 10.1016/j.parkreldis.2008.07.005. Epub 2008 Aug 23. PMID: 18723384; PMCID: PMC2693935.
- Immagini tratte da Holzbaur EL, Scherer SS. Microtubules, axonal transport, and neuropathy. N Engl J Med. 2011 Dec 15;365(24):2330-2. doi: 10.1056/NEJMcibr1112481. PMID: 22168648; PMCID: PMC3776444 e Tsuboi Y, Mishima T, Fujioka S. Perry Disease: Concept of a New Disease and Clinical Diagnostic Criteria. J Mov Disord. 2021 Jan;14(1):1-9. doi: 10.14802/jmd.20060. Epub 2020 Sep 21. PMID: 32942840; PMCID: PMC7840237.