Le miopatie infiammatorie idiopatiche (IIM), anche conosciute come miositi, rappresentano un gruppo eterogeneo di patologie muscolari accomunate dalla stessa base fisiopatologica: una infiammazione cronica a carico del muscolo scheletrico; spesso si tratta di condizioni sistemiche, con coinvolgimento di altri distretti: cute, sistema respiratorio e cardiovascolare, tratto gastrointestinale, articolazioni. Nel corso degli anni si è tentato di ideare un sistema classificativo adeguato alle molteplici manifestazioni cliniche; questo comprende una combinazione di caratteristiche cliniche, sierologiche ed istopatologiche.
L’esigenza di classificare trovò per la prima volta esito nel 1970 con i criteri di Medsger et al. Successivamente Bohan & Peter nel 1975 hanno rivisitato tali criteri diagnostici proponendo i seguenti sottogruppi: polimiosite, dermatomiosite, dermatomiosite giovanile, sindromi da overlap (miosite + altra condizione reumatologica associata), miosite associata al cancro.
In questi criteri, tuttavia, non veniva menzionata la miosite a corpi inclusi (IBM) come sottogruppo separato. Inoltre, tra le manifestazioni polmonari, non era annoverata la pneumopatia interstiziale, tipica di alcune miositi. Nel corso degli anni, la scoperta di determinate caratteristiche istopatologiche di alcune miositi ed il riscontro di numerosi autoanticorpi associati (MAAs) e specifici (MSAs), ha consentito lo sviluppo di nuovi criteri classificativi, i più recenti dei quali sono quelli dalla EULAR-ACR del 2017 per le IIM dell’adulto e giovanili (si veda la tabella di seguito).
Questi criteri prevedono due possibilità: casi in cui sia stato possibile eseguire una biopsia muscolare e casi nei quali tale procedura non sia stata eseguita per i più disparati motivi.
CRITERI EULAR-ACR (2017)
- BIOPSIA MUSCOLARE NON DISPONIBILE
- Probabile IIM: score 5.5-7.5
- Definita IIM: score >7
- BIOPSIA MUSCOLARE DISPONIBILE
- Probabile IIM: score 6.7-8.7
- Definita IIM: score >8.7
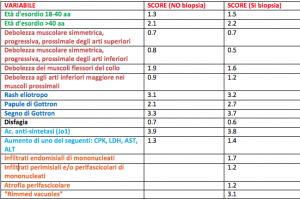
Ciò rende così l’esame istopatologico un fattore aggiuntivo di rilevanza elevata ma non necessariamente dirimente per la diagnosi di miosite. Vengono considerati una serie di fattori cui si attribuisce un punteggio; la somma dei punteggi ottenuti per ciascuna voce comporterà uno score finale: sia nel caso in cui la biopsia muscolare sia stata eseguita che nei casi in cui non sia disponibile un esame istopatologico, in base allo score ottenuto vengono riconosciute situazioni in cui una IIM è ritenuta probabile (probabilità compresa tra il 55 e il 90%) o definita (probabilità>90%). Il punteggio più elevato deriva dalla presenza di anticorpi anti-sintetasi. Tra i vari vantaggi, questi criteri permettono che non sfugga la diagnosi di alcune condizioni come la “dermatomiosite amiopatica” non associata a debolezza muscolare ma in cui sono presenti manifestazioni cutanee tipiche della dermatomiosite, insieme alla presenza di autoanticorpi.
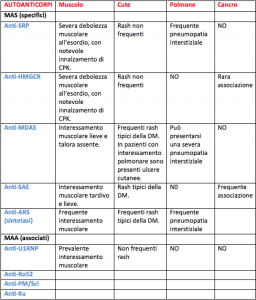
Per quanto concerne gli autoanticorpi, essi vengono classificati in due sottogruppi: myositis-specific autoantibodies (MSAs) e myositis-associated autoantibodies (MAAs). I MSAs sono specifici di determinate forme di miosite e sono prevalentemente correlati a manifestazioni extramuscolari (per esempio gli anticorpi anti-sintetasi sono fortemente associati a pneumopatia interstiziale e ad un decorso sfavorevole), mentre i MAAs possono essere riscontrati in altre condizioni reumatologiche che si possono sovrapporre alla miosite (es: LES, SSc…).
Una forma emergente di miosite idiopatica è la miopatia autoimmune necrotizzante (NAM), associata strettamente a due tipologie di autoanticorpi: ab anti-SRP (signal recognition particle) e ab anti-HMGCR. Dal punto di vista istopatologico si reperta necrosi delle fibre muscolari con minimi o assenti infiltrati flogistici. I livelli di creatinchinasi sono estremamente elevati. I pazienti presentano una notevole debolezza muscolare, spesso associata a mialgie, scarsamente responsiva ai trattamenti immunosoppressivi convenzionali. Gli ab anti-HMGCR si repertano frequentemente (ma non sempre) in pazienti che hanno fatto uso o che stanno assumendo statine; va infatti ricordato che tali autoanticorpi possono essere presenti anche in pazienti che non assumono terapie ipocolesterolemizzanti, in quanto delle statine naturali sono presenti anche in alcuni alimenti (es. funghi, mele ecc..). Infine, in pazienti che fanno uso di statine ma che non presentano sintomi muscolari o innalzamento di CPK sono stati riscontrati auto-anticorpi anti- HMGCR.
BIBLIOGRAFIA:
“Classification of myositis” – I. Lundeberg et al. – 2018 – Nature reviews rheumatology
“Autoantibodies in myositis” – N. McHugh et al. – 2018 – Nature reviews rheumatology
“Anti-HMGCR Autoantibodies in European Patients with Autoimmune Necrotizing Myopathies. Inconstant Exposure to Statin” – Y. Allenbach et al. – 2014 – Medicine Journal
-“Immune-Mediated Necrotizing Myopathy: Update on Diagnosis and Management” – P. Basharat et al. – 2015 – Curr. Rheumatol Rep
Stefania Martina Angelocola (Università Politecnica delle Marche – Ancona)