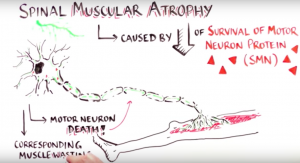
La storia naturale della SMA è complessa e variabile. Sono stati definiti 4 sottotipi clinici sulla base delle tappe motorie raggiunte durante lo sviluppo e dell’età d’esordio. Caratteristica comune a tutte le forme è l’atrofia muscolare e il pattern di coinvolgimento muscolare, tipicamente prevalentemente prossimale e simmetrico.
La SMA 0 riconosce un esordio prenatale (ridotti movimenti fetali), grave ipotonia e debolezza muscolare alla nascita. I neonati manifestano areflessia OT, diplegia facciale, difetti del setto interatriale. L’aspettativa di vita è di qualche settimana e generalmente la morte sopraggiunge per insufficienza respiratoria. La SMA 1 si caratterizza per ipotonia (aspetto delle gambe a “rana”, scarso controllo del capo), ipo/areflessia, debolezza muscolare con respiro addominale (per il relativo risparmio del diaframma rispetto ai muscoli intercostali). Per definizione i bambini con SMA1 non raggiungono mai la capacità di sedersi senza aiuto. L’esordio è entro i 6 mesi e l’aspettativa di vita inferiore ai due anni. I bambini con SMA 2 raggiungono la capacità di sedersi senza aiuto, ma non riusciranno mai a camminare autonomamente. In questa forma, con esordio tra i 6 e i 18 mesi, le principali comorbilità sono ortopediche (scoliosi, anchilosi), dovute alla progressiva debolezza muscolare. L’aspettativa di vita varia dai 10 ai 40 anni. Nella SMA 3 i bambini raggiungono la capacità di camminare autonomamente. L’esordio è generalmente dopo i 18 mesi e l’aspettativa di vita sovrapponibile a quella della popolazione generale per lo scarso coinvolgimento respiratorio. Nel caso in cui l’esordio della sintomatologia avvenga in età adulta si parla di SMA 4. La gravità clinica della SMA correla inversamente con il numero di copie del gene SMN2.
Responsabile della malattia sono mutazioni a carico del gene SMN1, principale responsabile della sintesi della proteina SMN (Survival Motor Neuron protein), che va incontro a deplezione. Una minor percentuale di SMN protein viene sintetizzata a partire dal gene SMN 2, gene quasi identico a SMN 1, ma che differisce da questo per alcuni nucleotidi tra cui un nucleotide specifico nell’esone 7, che causa un’alterazione di splicing a livello del pre-mRNA. Questo fa sì che solo il 10% della proteina full-lenght venga sintetizzata a partire dal gene SMN2, mentre il restante 90% è un trascritto mancante dell’esone 7 (Δ7SMN) non funzionale. Nella SMA, quindi, bloccando la sintesi di SMN a partire dal gene SMN1, resta esclusivamente la quota di SMN prodotta a partire dal gene SMN2. ”
Recentissima e promettente novità è rappresentata da Nusinersen (Spinraza), un oligonucleotide antisenso. In questo canale youtube si vedono dei genitori di una bambina affetta da SMA che ha avuto accesso al farmaco in America e che hanno documentato le varie infusioni e le tappe raggiunte dalla bambina.
Tale farmaco va ad interferire con la sintesi della proteina SMN2 a livello post-trascrizionale. Lega il pre-mRNA del gene SMN2 a livello di un sito ISS-N1 (Intronic Splice Silencing) presente nell’introne 7. Attraverso tale legame Nusinersen sposta i fattori di splicing, che normalmente inducono eliminazione del’introne 7 e facilita l’inclusione di tale introne nell’mRNA. In tal modo, c’è maggior probabilità che a partire da tale mRNA venga generata la proteina SMN funzionante full lenght, compensandone il deficit. Ad oggi tale farmaco è stato approvato in Italia e reso disponibile per tutti i malati di SMA.
Alessandro Introna, Bari
Alessandro Bombaci, Torino
Paola Alberti, Milano Bicocca